
How to solve a jigsaw puzzle with 1.56 billion pieces
– or the challenging route to giga-scale benchmarking datasets for machine learning-accelerated drug discovery
Drug discovery efforts typically feature fast computational screening methods to find small organic molecules binding to a drug target, e.g., an enzyme critical to pathogen survival. The target can be envisioned as a partially completed biological jigsaw puzzle in 3D.
In this analogy, computational docking would represent a method that tries to fit every single piece among hundreds of candidates into the existing jigsaw frame. How well shape and pattern match is translated into a ‘docking score’ by which docking predicts the best fit.
The catch of a biological jigsaw? There is no perfect match! Hence, drug discovery projects typically rely on thousands of diverse small molecules to find a few that match the desired overall picture as well as possible and thus score best.
Predicting docking scores with Machine Learning
Recently, this brute-force nature of docking, trying piece after piece one-by-one, has been put in jeopardy by the size of available small molecule collections: With modern compound libraries exceeding the billion scale, docking would suddenly take months or years, even with supercomputing resources at one’s disposal.
But what if there were a faster way to predict a piece’s fit? This is where machine learning (ML) comes in: When presented with enough examples and their corresponding scores, ML can learn what makes some pieces fit well and others not. Predicting docking scores with such a model is then much faster than conventional docking methods.
Unsurprisingly, the acceleration of docking studies with ML to keep up with modern giga-scale compound libraries has become a popular approach – with a catch: A prediction is hardly ever perfect and thus, some of the best pieces might be lost. If you developed a novel method to speed up docking, users of your method would likely want to know how many – and so did we.
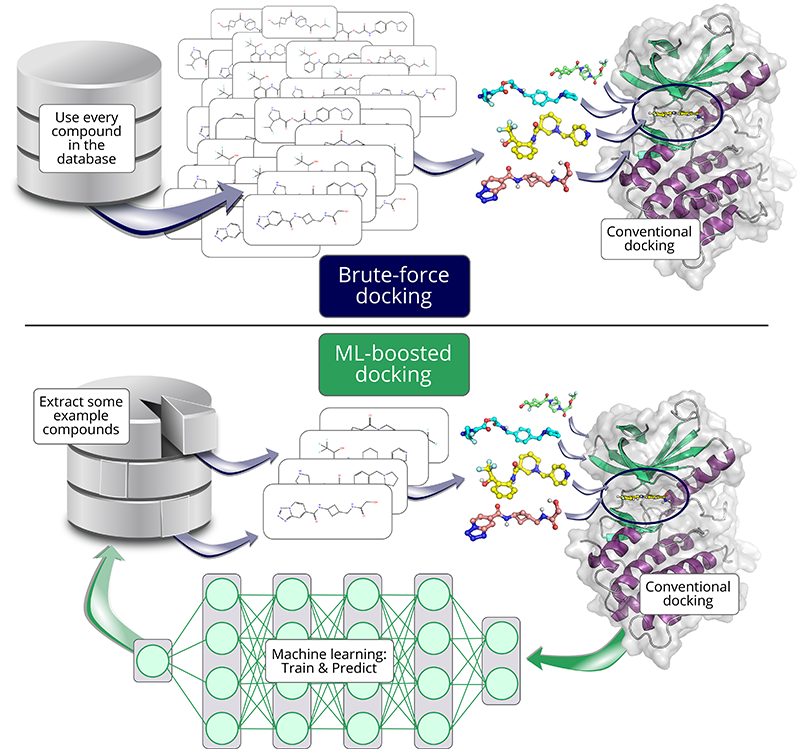
One tool to speed up docking studies with the help of ML is HASTEN. HASTEN had been previously shown to successfully increase the speed of docking millions of compounds, but how would the tool fare on the giga-scale?
It is hardly surprising that just like HASTEN prior to our study, most tools have not actually been benchmarked on giga-scale data: Comparing ML approaches with brute-force docking results first requires the availability of a ‘ground truth’: All the scores for all the billions of pieces need to be known to determine which ones would not be found by the ML model.
From 4 months to 10 days
To rigorously determine how many promising compounds we would be missing and how much time we could save, we set out to generate brute-force docking results of 1.56 billion compounds against an anti-bacterial and an anti-viral target – and compare those with the results obtained by the ML-boosted HASTEN.
Although we used the particularly fast Schrödinger Glide HTVS method, we were still facing several months of docking studies. Glide, as most other docking tools, has been developed with more traditional library sizes in mind and on this unprecedented scale, we faced a number of practical challenges, such as splitting the task at hand into thousands of jobs to fit into the CSC walltime limits while keeping track of the progress, or aspects of resource management – just imagine your table is meant to hold a 1000-piece jigsaw and you suddenly attempt to make it hold 1 000 000 000 pieces…
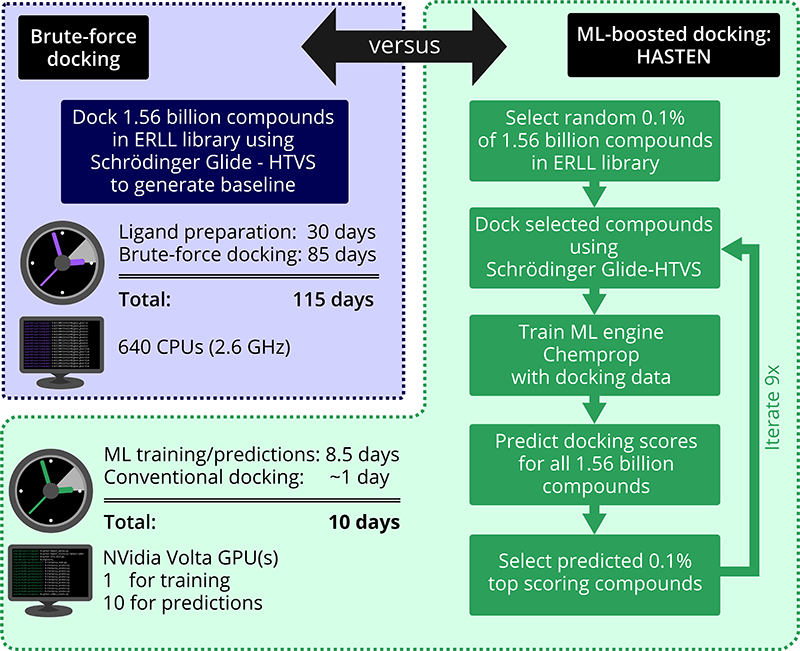
Our project was able to show that tools like HASTEN can bring down the time for screening more than one billion compounds from almost 4 months to around 10 days. And not only that: Using the ML-boosted approach, in a fraction of the time, we still got more than 90% of the very top-scoring compounds that were found in the brute-force docking.
The time requirement and the resource-related challenges we faced in this project when generating a docking ground truth for benchmarking HASTEN are the key reason why many similar tools and approaches have not actually been validated/tested on the giga-scale. Since more performant methods are crucial to the future utilization of billion-scale compound libraries and libraries keep growing, novel methods are continuously developed and need appropriate benchmarking on the intended use-scale. To that end, we released our entire 1.56 billion compound-docking results for two targets as benchmarking data into the public domain. We believe that the availability of such datasets is crucial for the advancement of the entire field and a long-term investment into state-of-the-art screening methods that save time and resources – for everyone.
Read more:
- Pre-print ChemRxiv: Machine Learning-Boosted Docking Enables the Efficient Structure-Based Virtual Screening of Giga-Scale Enumerated Chemical Libraries, Toni Sivula, Laxman Yetukuri, Tuomo Kalliokoski, Heikki Käsnänen, Antti Poso and Ina Pöhner. (https://doi.org/10.26434/chemrxiv-2023-g34tx)
Ina Pöhner
The author is a Postdoctoral Researcher in the Molecular Modeling and Drug Design Research group at University of Eastern Finland. She loves taking on data-intensive challenges in drug discovery projects and she solved this jigsaw puzzle in close cooperation with CSC specialists.
Efficient deployment of high-throughput virtual screening workflow
CSC worked closely with UEF for the efficient deployment of a high-throughput virtual screening workflow using Schrödinger’s Maestro software.
The built-in workflow automation in Schrödinger has been designed for smaller data sets and scaling it up required detailed analysis of the individual steps and their overall impact on the computing platform. Simply scaling up would have been inefficient and would have interfered with other users. At this stage we also decided to move to Mahti to access more resources, which also required a different setup to launch jobs.
The porting and optimization of the workflow involved planning the dataset partitioning also with the later usage and publishing in mind, trimming unnecessary steps, tuning the built-in parameters, optimizing the parallel distribution, planning optimal license usage and wrapping all of this in a robust script.
One of the critical changes was to bypass heavy disk I/O operations by using memory instead. In addition to speeding up the workflow, it minimized the interference with other users. Many of these steps required deep understanding of the Mahti infrastructure and access to system monitoring data. The extra CSC effort is justified by the efficient usage, i.e. more resources to other researchers, as well as the increased knowledge of workflows to be used in support of others.
Atte Sillanpää and Laxmana Yetukuri, CSC