
Efficient molecular dynamics simulations on LUMI
The main computing power of the LUMI supercomputer comes from its hefty GPU partition, LUMI-G, composed of 10 240 AMD MI250X graphics processing units. The high performance of these latest generation accelerators has already been proven for use cases such as natural language processing and astrophysical hydrodynamics, but how does the performance look like for molecular dynamics? Spoiler alert: it’s excellent!
Classical molecular dynamics (MD) enables atomic-scale simulations of biomolecular systems such as proteins and is one of the most used methods in CSC’s computing environment.
A popular simulation code that implements MD is the GROMACS open source software package, which is especially known for its efficiency and flexibility. While GROMACS has had excellent support for Nvidia GPUs through the CUDA programming model for a long time, support for AMD GPUs has only recently matured following developments in the OpenSYCL backend (formerly known as hipSYCL) which GROMACS uses to enable GPU acceleration on AMD hardware.
Our experience with GROMACS on LUMI-G shows that large systems composed of several hundred thousand to millions of atoms are able to scale up to full LUMI-G nodes very well. The figure below compares the performance of GROMACS 2023.1 on LUMI-G to that on CPUs on the Mahti supercomputer.
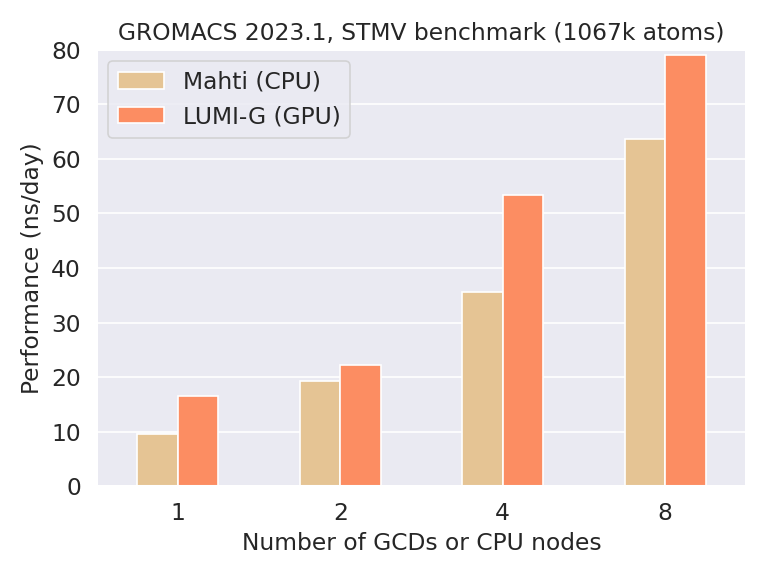
The studied benchmark system is a satellite tobacco mosaic virus (STMV) consisting of ca. 1 million atoms in total. Besides the good scaling, the figure shows that already a single AMD MI250X graphics compute die (GCD), which corresponds to half a GPU, outperforms one Mahti CPU node of 128 cores. This demonstrates nicely the performance of the cutting-edge accelerators available on LUMI and should be taken as a rough guideline regarding performance expectations when migrating simulations to LUMI. In other words, if your simulation is much slower on one MI250X GCD compared to one Mahti CPU node, there’s probably something in your submission script settings that should be improved.
A common source of slow performance on LUMI-G is that reserved CPU cores are not bound to the correct GPUs. Why this matters is because certain cores on a LUMI-G node are always directly linked to a specific GPU and disregarding this binding may result in substantial communication overhead. To ensure appropriate CPU affinity, refer to our script template in Docs CSC!
Speed vs. throughput
The previous benchmark shows that medium-sized and large use cases are more than suitable to be run at scale on LUMI-G. But what about smaller systems involving, say, ten to one hundred thousand atoms? Running such simulations on a single GCD can be reasonably efficient, but the smaller a system gets, the poorer it will be able to leverage the full capacity of the accelerator.
Fortunately, the MI250X GPUs on LUMI-G have native support for running multiple MPI ranks per GPU similar to the multi-process service (MPS) feature of Nvidia GPUs. In other words, if you are able to split your MD use case into multiple independent simulations, sharing a smaller set of GPUs between these is an easy way to improve the GPU utilization. Typical examples include enhanced sampling techniques as well as high-throughput multi-simulations where differently equilibrated copies of a system are simulated concurrently for increased aggregate sampling across multiple trajectories.
GROMACS implements multi-simulations via the built-in multidir feature, which shares the allocated computational resources equally among all input systems. Using a fixed number of two LUMI-G nodes (16 GCDs), the benchmark below demonstrates the effect of varying the number of trajectories per GCD on the overall throughput.
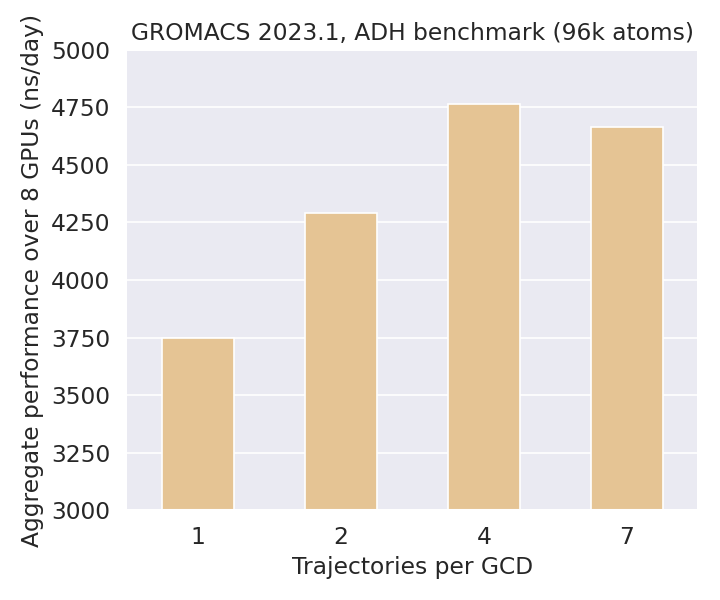
For the benchmarked alcohol dehydrogenase (ADH) enzyme (ca. 96 000 atoms), increasing the number of trajectories per GCD from one to four improves the aggregate performance by roughly one microsecond/day owing to better GPU utilization. While this benchmark was run on 2 GPU nodes, one could easily scale it to an arbitrary number of nodes due to the simulations being completely independent of each other.
For example, a total combined throughput of 100 microseconds per day could be accomplished using 42 LUMI-G nodes, which is less than 2% of the total capacity of the GPU partition. Reaching a similar performance on Mahti with one trajectory per 32 CPU cores would require a resource allocation of roughly 560 nodes (40% of Mahti) – a tangible demonstration of the unprecedented computational capacity of the LUMI supercomputer!
Take-home messages:
- Most systems run well on a single GCD, performance typically better than on one 128 core CPU node
- Large systems (several 100 000 – 1M atoms) are usually able to scale to multiple GPUs (mind the proper binding of CPUs and GPUs)
- GPU utilization can be maximized for small systems (less than 100 000 atoms) by running multiple independent trajectories per GCD (multidir feature)
- Don’t hesitate to contact CSC Service Desk (servicedesk@csc.fi) if you need help in migrating your simulations to LUMI!
Further reading:
- Create a LUMI project and apply for GPU resources
- Using GROMACS on LUMI-G
- High-throughput computing with
- GROMACS LUMI documentation
Rasmus Kronberg
Author works in CSC’s science support as a computational chemistry specialist.